





Neurodegeneration
A complication of long- standing multiple sclerosis (MS)
Abstract
Approximately 85% of patients with multiple sclerosis are first affected by relapsing and remitting disease, with periods of acute exacerbation. The majority of these patients also develop a secondary course of disease, characterized by progressive, gradual loss of function. These two types of disease processes appear to be driven by independent mechanisms, and clinicians should address not only the underlying inflammatory stimuli to reduce periods of exacerbation, but also address targets of chronic neurodegeneration. This article focuses on four key areas with regard to neurodegeneration: mitochondrial dysfunction, cerebrovascular function, iron deposition, and excitotoxicity, to prevent the onset of or decrease the severity of progressive MS.
Over the past five years (2020–2025), one of the most significant advances has been the clinical validation of Bruton’s tyrosine kinase (BTK) inhibitors, which offer promising potential to slow neurodegenerative progression in progressive MS. Notably, tolebrutinib has demonstrated a 31% delay in disease progression in non-relapsing secondary-progressive MS during Phase III trials. Concurrently, fenebrutinib has shown strong suppression of MS activity and disability progression, including early evidence of brain penetration and microglial modulation, with durable effects observed in relapsing and primary progressive MS cohorts. Additionally, emerging research has revealed that neurons within MS lesions accumulate DNA mutations at over twice the rate of healthy neurons, implicating inflammation-driven genetic instability as a contributor to neurodegeneration and opening new therapeutic avenues. These discoveries represent a shift from solely managing inflammatory relapses to also addressing the core drivers of neurodegeneration in long-standing MS.
Introduction
It is well accepted that multiple sclerosis (MS) is a Th1/Th17-mediated autoimmune disease (Korn 2008). In MS about 85% of patients initially experience a relapsing-remitting course of disease (relapsing-remitting multiple sclerosis, RRMS), which is characterized by acute episodes of neurological deficit called “relapses.” These episodes commonly include limb weakness, vision changes, ataxia and sensory disturbances. Relapses are followed by periods of remission, which usually have partial or near-complete restoration of function.
After a variable number of years, the majority of these patients develop a secondary progressive disease course where disability slowly accrues despite often having fewer attacks, suggesting that other mechanisms are involved in progression. Furthermore, commonly used anti-inflammatory medications such as interferon beta have minimal effect on inhibiting the neurological decline, implying that this process is more complex than inflammation alone (Su 2009). Latency to the commencement of progressive MS varies greatly. One study demonstrated a broad range from one to 36 years with a mean of five years in the quickest 25% to greater than 15 years in the slowest 25% (Scalfari 2013). Male sex, older age of onset (>30), and high early yearly relapse frequency (>3) were predictors for more rapid entry into progressive MS (Scalfari 2013). Therefore, because some patients may convert quickly into progressive MS, preventing progressive MS should be a primary goal when treating all MS patients, especially if they are at higher risk.
Relapses are considered to be the clinical manifestation of acute inflammatory demyelination in the CNS, and progression of disability is thought to reflect chronic demyelination, axonal loss and neuronal death (Luessi 2012). MS should be viewed as both an inflammatory and a neurodegenerative condition, which has major implications for therapy; in particular, there is a need for ongoing CNS protection in addition to controlling acute inflammatory exacerbations.
Treatment of Progressive MS
In an attempt to reduce relapse frequency and severity in RRMS patients, I typically employ renditions of the strategies suggested in Table 1. However, it is essential to include neuroprotective strategies in addition to a solid anti-inflammatory, relapse-oriented protocol to delay disease conversion to progressive MS. The following discusses a primarily theoretical approach to reducing the onset and severity of progressive MS by looking at some of the most prominent underlying mechanisms. Unfortunately, many of these have not been studied in humans with the specific endpoint of slowing disability progression. However, I have chosen to present the options that, in my opinion, are theoretically sound for clinical use. I will focus on four key areas that I emphasize in my practice with regard to neurodegeneration: mitochondrial dysfunction, cerebrovascular function, iron deposition, and excitotoxicity, in order to prevent the onset of or decrease the severity of progressive MS.
Mitochondrial Dysfunction
Mitochondrial dysfunction in MS may be mediated by the pathological opening of the mitochondrial permeability transition pore (PTP) located in the inner mitochondrial membrane. The PTP is a channel that activates and opens in response to mitochondrial stress (Su 2013).
In MS, loss of myelin greatly impairs the efficiency of action potential propagation, resulting in mitochondrial dysfunction and damage. The chain of events proceeds: first, in response to demyelination, sodium channels become redistributed along the axon, and their synthesis is upregulated. This results in increased energy requirements to maintain neuronal function. When the demand for ATP exceeds the production capabilities of existing mitochondria, the Na+/K+ ATPases begin to fail. An excess of Na+ ions accumulates within the intracellular space, and eventually this reverses the Na+/ Ca2+ exchanger that normally moves Na+ into the cell and Ca2+ from the cell. Instead, calcium accumulates within the cell. The PTP pore then opens in response to elevated calcium; however, persistent PTP opening leads to loss of the mitochondrial membrane potential, equilibration of ionic gradients, and promotes mitochondrial matrix swelling and outer membrane rupture (Su 2009).

targeting relapse severity and frequency
Treatments that increase ATP production may prevent PTP dysfunction. We can attempt to increase ATP production with the use of creatine monohydrate. It is well known that exogenous creatine supplementation provides additional phosphocreatine, which acts as a reserve of high-energy phosphates for ATP production (Rosenfeld 2008). Creatine is taken up by the neuron via specific creatine transporters and phosphorylated to the high-energy phosphocreatine by either mitochondrial or cytosolic creatine kinases (Adhihetty 2008).
Supplying exogenous creatine monohydrate can provide additional substrate for ATP production to meet the increased demand that occurs with demyelination. The activity of creatine monohydrate as a nootropic agent has been demonstrated in healthy adults by supplementing with 5g per day for six weeks, resulting in improvements in working memory (backward digit span) and intelligence (Rae 2003). Creatine has also been shown to maintain the mitochondrial creatine kinase in an octameric conformation. Mitochondrial creatine kinase can exist as either a dimer or an octamer, and the enzyme’s function is determined by its structure (Adhihetty 2008). The octameric form of the mitochondrial creatine kinase interacts with components of the PTP to suppress pore opening and potentially reduce mitochondrial apoptotic susceptibility (Adhihetty 2008). On the other hand, upon exposure to oxidative stress, the enzyme undergoes a conformational change to the dimeric form, losing this functionality (Adhihetty 2008). A “slow load” of creatine supplementation for 28 days at a rate of 3g/day may be considered equivalent to a six-day “fast load” protocol of 20g per day used in athletes, both with a 2g per day maintenance dose thereafter. Based on this, I typically recommend one-half teaspoon for month one, with maintenance of approximately one-third of a teaspoon. Creatine supplementation is easy and convenient as it rapidly dissolves in water and is extremely cost-effective for long-term use.
Acetyl-L-carnitine (ALCAR) is another promising substance that enhances mitochondrial function by facilitating fatty acid transport. To enter the mitochondria, fatty acids must bind to coenzyme A, forming fatty acyl-CoA. Long-chain fatty acyl- CoA molecules are too large to cross the internal mitochondrial membrane and rely on enzymatic transportation that requires L-carnitine (NMCD Carnitine monograph). ALCAR administration results in increased intracellular levels of L-Carnitine. Both IV and oral administration of acetyl-L-carnitine result in a corresponding increase in cerebrospinal fluid concentrations of ALCAR, indicating it readily crosses the blood-brain barrier and can facilitate delivery of additional substrate for ATP synthesis to mitochondria (Alt Med Rev 2010).
ALCAR has been studied clinically in MS patients. Evidence of improved fatigue (implying improved neuronal function) was seen in 36 MS patients who were treated for three months with either amantadine (pharmaceutical for MS fatigue) or ALCAR (1g twice daily); after a three month washout period, the patients were crossed over to the opposite treatment. The authors found that 29% of patients improved after ALCAR compared to 21% after amantadine (Tomassini 2004).
With regards to acute inflammation, ALCAR may also decrease reactive nitric oxide (NO) derivative species. Reactive nitric oxide derivatives are cytotoxic to oligodendrocytes and neurons in culture by inhibiting the mitochondrial respiratory chain. MS patients are known to have increased nitrosative stress, as activated glia secrete reactive nitrogen species (Bizzozero 2005). In a clinical trial, ten MS patients were treated for six months with 2g ALCAR and compared to untreated MS subjects or patients with non-inflammatory neurological conditions. Prior to treatment, concentrations of reduced glutathione were approximately 38% lower in MS patients compared to controls, implying increased baseline inflammatory activity. Treatment with ALCAR resulted in decreased CSF levels of NO reactive metabolites as well as increased content of reduced glutathione (Calabrese 2003).
In my experience, the effects of ALCAR on MS fatigue are subtle, however, it is an extremely well tolerated intervention, and provides acute inflammatory as well as chronic metabolic support. A dose of 1.0- 1.5g bid is a common recommendation.
Dosing on an empty stomach is recommended, to maximize its absorption in the jejunum (Alt Med Rev 2010).
Cerebrovascular Function
CCSVI (cerebrospinal venous insufficiency) is an area that MS patients often inquire about. CCSVI refers to the idea that blocked extracranial venous blood outflow causes cerebral venous reflux in MS patients. It seems now that scientific evidence supporting a causal relationship between CCSVI and MS is lacking. If MS is associated with CCSVI, it is most probably an acquired phenomenon that occurs particularly in MS patients associated with age (Lanzillo 2013). Nonetheless, in my practice I have observed substantial improvements in neurological function in some patients following CCSVI surgery, and from a non-surgical perspective, it is well known that cognition improves among non-MS patients following use of naturally occurring nootropics that improve blood flow, such as ginkgo. Ultimately, whether or not impaired blood flow is an etiological phenomenon in MS, improving cerebral circulation remains an important area of focus when addressing neurodegeneration; improved circulation results in improved oxygen and nutrient delivery to neurons experiencing increased energy demands. The two agents I use often are Ginkgo biloba and Vinpocetine.
In MS patients, ginkgo has been shown to improve both functional and cognitive parameters when dosed at 240mg per day (Johnson 2006, Lovera 2007). The Johnson study used an extract standardized to 24% flavonoid glycosides, 6% terpene lactone (2006), while the Lovera study used an extract standardized to 31.4% flavonoids, 4.5 % terpenes (2007). Neuroprotective effects of ginkgo have been demonstrated in several in vitro and in vivo models, specifically showing protection of cultured neurons against death induced by hypoxia, glutamate and nitric oxide (Ahlemeyer 2003). Ginkgo also appears to have IL-6 lowering ability when given to patients with age-related neurological disorders (Ching-Hsiang 2012). Interestingly, ginkgo appears to be able to increase endothelial nitric oxide synthase (eNOS) mediated NO production, resulting in vasodilation, whereas it inhibits inducible NOS (iNOS) mediated NO production, thereby preventing excessive NO synthesis by macrophages (Ahlemeyer 2003). This suggests that Ginkgo may reduce acute inflammation while providing increased blood flow to damaged neurons. My preference has been to dose Ginkgo at 120mg bid (standardized to 24% and 6%) because it’s readily available at this standardization, and reflects the dose used in Johnson (2006).
Vinpocetine is an interesting substance due to its ability to increase blood flow, as well as its therapeutic potential in neurogenic bladder. In one study, intravenous vinpocetine increased both global and regional cerebral blood flow, with 36% and 37% increases in blood flow to the thalamus and caudate nucleus respectively, as measured by positron emission tomography (PET) (Szilágyi 2005). An oral dosing study using PET imaging also demonstrated increased cerebral blood flow as well as improvements in cognition in patients with mild cognitive impairment due to cerebral hypoperfusion (Valikovics 2007).
Vinpocetine has also been studied for bladder dysfunction. In a pilot study of 19 patients with urge incontinence, sensory incontinence and low compliance bladders, three patients reported slight improvement, and eight reported pronounced improvement within four weeks (Truss 2000). MS patients with neurogenic bladder are considered motor urge incontinent (overactive detrusor). A larger follow up trial in a “worst case scenario population” with primarily detrusor instability showed less pronounced benefits (Truss 2001). Both the vascular and bladder related improvements are attributed to vinpocetine’s phosphodiesterase-1 inhibitory activity. Viagra is a pharmaceutical phosphodiesterase-5 inhibitor (Truss 2001). Phosphodiesterase inhibitors (PDEIs) suppress TNF-alpha production by various cells and suppress experimental demyelination. In a pilot study of 12 MS patients, a combination of three PDEIs was shown to reduce annual relapse rates (Suzumura 2000). Under fasting conditions, vinpocetine has about 7% bioavailability so it must be given with food (Paytar 2011). I typically dose 45mg per day, however 30-60mg is suggested by a 2003 Cochrane review as being suitable for cognitive impairment (Szatmari 2003). Emprical use of vinpocetine for bladder function should be for at least one month, as results were seen within one month in the study by Truss (2000). Precautions related to risk of bleeding relate to both ginkgo and vinpocetine (NMCD Vinpocetine monograph).
Iron Deposition
Zamboni’s CCSVI theory proposed that venous reflux leads to iron deposition, inflammation and leukocyte infiltration. Regardless of whether the CCSVI hypothesis is correct, iron is in fact implicated in a number of neurodegenerative diseases and senile dementia since it accumulates in the brain with age, and ionic iron can in turn participate in the Fenton reaction with subsequent generation of ROS, initiating the processes of oxidative stress (Singh 2009, Weinreb 2009).
In one study, the brain tissue of 33 MS and 30 control cases were analyzed. In active MS lesions, iron was apparently released from dying oligodendrocytes, resulting in extracellular accumulation of iron and it was suggested that cellular degeneration in MS lesions leads to waves of iron liberation, which may further propagate neurodegeneration (Hametner 2013).
In mice being fed oral EGCG, a human equivalent dose of three litres of green tea, dramatically suppressed experimental autoimmune encephalomyelitis (EAE, the animal model of MS) (Atkas 2004). EGCG inhibits Th1 and Th17 differentiation, NF-kb, and ROS, making it one of my top choices for use in reducing relapse severity and frequency (Wu 2012). However, in the context of neurodegeneration, EGCG can also chelate ionic iron to form inactive complexes (Weinreb 2009).
Interestingly, a recent study has shown in-vivo synergy of EGCG and copaxone (daily injectable immune-modulating MS treatment) in EAE, and the authors encouraged the combination of anti-inflammatory and neuroprotective treatments (Herges 2011). Converting the mouse EGCG dose (300 ug bid) used in the first EAE study to an equivalent human dose (assuming 60kg human and 20g mouse) yields approximately 1459mg EGCG as the therapeutic human dose (Mouse-Genome 2010, Reagan-Shaw 2008).
Although there are many factors to be considered in extrapolating animal dosages and their effects to humans, this study does
provide a loose estimate. In practice, I dose EGCG at about 900mg per day when trying to reduce relapse frequency or as an adjuct to interferon, since dosing of EGCG alone at 800mg is a dose that has consistently been shown to be safe in a human pharmacokinetic study. There have been case reports of hepatotoxicity with EGCG, which suggest some caution should be used with higher dosages. The other catechins present in green tea extract possess similar properties to EGCG, and can be used to help make up the full catechin dose used in the EAE study (Chow 2003, Shanafelt 2009). In terms of using EGCG to prevent neurodegeneration, I encourage, at the very least, daily ad libitum consumption of green tea beverage to ensure some degree of iron chelation as part of their neuroprotective regime.
Excitotoxicity
Evidence has accumulated that excessive glutamate is released at the sites of demyelination and axonal degeneration in MS plaques, and the most probable candidates for this cellular release are infiltrating leukocytes and activated microglia (Frigo 2012). One study looking at MS patients compared to healthy patients, found the presence of elevated glutamate in active lesions using 3D MRI. One of the primary glutamate receptors, the NMDA receptor, allows the influx of cations, though most notably calcium. Excessive glutamate stimulation causes excessive intracellular calcium accumulation, leading to excitotoxic injury (Lau 2010). One study looked at 16 patients with MS, where one year without treatment was followed by 1-year riluzole (inhibits glutamate release from neurons). Treatment resulted in reduced spinal atrophy and reduced T1 lesions (active lesions) (Killestein 2005). Although pharmacologic glutamate inhibitors are not currently being employed for this, we can use natural glutamate antagonists to combat the excitotoxicity. GABA receptors provide a counterbalance to glutamate receptors (Rossi 2012). My preference is to use daily magnesium as an adjunct to calcium supplementation; magnesium is a NMDA antagonist and GABA-A agonist, and I have seen good effects in MS spasticity with this approach (Held 2002). I always combine calcium with vitamin D prescription. As mentioned in Table 1, I often recommend 10,000 IU of vitamin D daily in MS. Studies of EAE and a 2010 human dose escalation study where doses reached 40 000 IU indicate that the immune modulatory actions of vitamin D may be contingent on concomitant calcium administration (approximately 1200mg) (Burton 2010, VanAmerongen 2004). As a result, I regularly suggest 10,000 IU vitamin D, 1200mg calcium, and 600mg magnesium nightly. The anxiolytics GABA and taurine can also be considered, especially if anxiety is part of the symptomatology, since taurine is a GABA agonist and may have independent neuroprotective effects, while GABA promotes specific patterns of brain activity (Abdou 2006, Oja 2007).
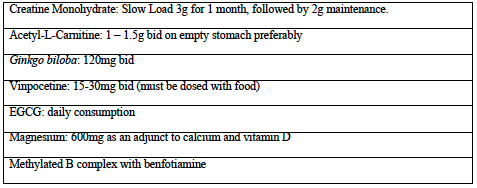
Lastly, as a foundation for more advanced supplement therapies, B-vitamins and a diet including daily berry consumption are often prescribed for general cognitive function. A B complex with methylated B12 and benfotiamine is preferred for homocysteine reduction and optimal nerve transmission. Lower median CSF vitamin B12 concentrations were found in groups of patients with MS and serum homocysteine is significantly increased in MS patients (Niist 1990, Vrethem 2003). Blueberry consumption has been shown to improve memory in older adults, has been shown to improve blood brain barrier integrity, and can suppress NF-kb by about 27% in humans when used at a dose of 200g daily (Karlsen 2007, Krikorian 2010, Robert 1977).
Promising prospective therapies that I may add to my formulary in future include: low dose lithium orotate, pyrroloquinoline quinone (PQQ), and Centella asiatica for their effects on brain derived neurotrophic factor (BDNF), nerve growth factor (NGF) and nerve regeneration respectively (Misra 2012, Rowe 2004, Soumyanath 2005). I look forward to more research emerging on the possible role of these agents.
In conclusion, MS should be treated as both an inflammatory and neurodegenerative condition. RRMS patients need neuroprotective support immediately, alongside their immune modulatory therapies, because the onset of progressive disease varies greatly and can even be immediate (primary progressive). It is my opinion that neuroprotective strategies addressing mitochondrial dysfunction, iron deposition, excitotoxicity, and vascular perfusion, in addition to anti-inflammatory treatments, are a well-rounded approach to reducing the possibility and severity of progressive disability accrual.
References
Abdou AM. Biofactors. 2006;26(3):201-8. Adhihetty PJ. Neuromolecular Med. 2008;10(4): 275-90
Ahlemeyer B. Cell Mol Life Sci. 2003 Sep;60(9):1779-92.
Aktas O. J Immunol. 2004 Nov 1;173(9):5794-800 Alt Med Rev. Altern Med Rev. 2010 Apr;15(1): 76-83.
Bizzozero OA. Neurochem Res. 2005 Jan;30(1): 139-49.
Burton JM. Neurology. 2010 Jun 8;74(23):1852-9. Calabrese V. Neurochem Res. 2003 Sep;28(9): 1321-8.
Chow HH. Clin Cancer Res. 2003 Aug 15;9(9):3312-9.
Ching-Hsiang L. Indian J Pharmacol. 2012 Jan;44(1):118-21.
Frigo M. Curr Med Chem. 2012;19(9):1295-9. Gelinas DF. Amyotroph Lateral Scler. 2008 Oct;9(5):266-72.
Greve B. J Neuroimmunol. 2001 Dec 3;121(1- 2):120-5.
Guggenmos J. J Immunol. 2004 Jan 1;172(1):661-8. Hadjivassiliou M. Lancet Neurol. 2010 Mar;9(3):318-30.
Hametner S. Ann Neurol. 2013 Jul 19.
Held K. Pharmacopsychiatry. 2002 Jul;35(4):135-43.
Herges K. PLoS One. 2011;6(10):e25456.
Johnson SK. Explore (NY). 2006 Jan;2(1):19-24.
Karlsen A. J Nutr. 2007 Aug;137(8):1951-4
Killestein J. J Neurol Sci. 2005 Jun 15;233(1-2): 113-5.
Korn T. J Neurol. 2008 Dec;255 Suppl 6:2-6.
Krikorian R. 2010 Apr 14;58(7):3996-4000.
Lau A. Pflugers Arch. 2010 Jul;460(2):525-42.
Lanzillo R. BMC Neurol. 2013 Feb 13;13:20.
Luessi F. Expert Rev Neurother. 2012 Sep;12(9):1061-76.
Lovera J. Mult Scler. 2007 Apr;13(3):376-85.
Mashayekh A. Neuroradiology. 2011 Mar;53(3):185-91.
Misra HS. J Biosci. 2012 Jun;37(2):313-25.
Mouse Genome Informatics. Accessed May 2 2010.
Nijst TQ. J Neurol Neurosurg Psychiatry. 1990 Nov;53(11):951-4.
NMCD Acetyl-L-Carnitine Monograph. Natural Medicines Comprehensive Database. Accessed online September 2013.
NMCD Vincpocetine Monograph. Natural Medicines Comprehensive Database. Accessed online September 2013.
Oja SS. Proc West Pharmacol Soc. 2007;50:8-15. Rae C. Proc Biol Sci. 2003 Oct 22;270(1529): 2147-50.
Rahn KA. Curr Med Chem. 2012;19(9):1335-45. Reagan-Shaw S. FASEB J. 2008 Mar;22(3): 659-61.
Robert AM. J Med. 1977;8(5):321-32. Rosenfeld J. Expert Rev Mol Med. 2004 Oct 18;6(21):1-18.
Rossi S. Mult Scler. 2012 Nov;18(11):1633-5. Santos RF. Pharmacopsychiatry. 2003 Jul;36(4): 127-33.
Scalfari A. J Neurol Neurosurg Psychiatry. 2013 Mar 13.
Shanafelt TD. J Clin Oncol. 2009 Aug 10;27(23):3808-14.
Shinto L. Prostaglandins Leukot Essent Fatty Acids. 2009 Feb-Mar;80(2-3):131-6. Singh AV. J Cereb Blood Flow Metab. 2009 Dec;29(12):1867-78.
Soumyanath A. J Pharm Pharmacol. 2005 Sep;57(9):1221-9.
Suzumura A. Mult Scler. 2000 Feb;6(1):56-8. Su KG. Curr Neurol Neurosci Rep. 2009 Sep;9(5):411-7.
Su K. Front Physiol. 2013;4:169. Szilágyi G. J Neurol Sci. 2005 Mar 15;229- 230:275-84.
Szatmari SZ. Cochrane Database Syst Rev. 2003;(1):CD003119.
Tomassini V. J Neurol Sci. 2004 Mar 15;218 (1-2):103-8
Truss MC. World J Urol. 2001 Nov;19(5):344-50.
Truss MC. World J Urol. 2000 Dec;18(6):439-43.
Valikovics A. Ideggyogy Sz. 2007 Jul 30;60(7- 8):301-10.
VanAmerongen BM. Eur J Clin Nutr. 2004 Aug;58(8):1095-109.
Vrethem M. Mult Scler. 2003 Jun;9(3):239-45.
Weinreb O. Genes Nutr. 2009 Dec;4(4):283-96.
Weinstock-Guttman B. Mol Aspects Med. 2012 Feb;33(1):107-18.
Yadav V. Mult Scler. 2010 Apr;16(4):387-97.








